Perfekte Daten zu Arzneimittelsicherheit oder Arzneimitteltherapiesicherheit/Pharmakovigilanz gibt es leider
nicht. Man steht vor der Wahl, welche Daten man nutzen will. Unterschiedliche Ansätze zu kombinieren ist
schwierig.
1. Ausgangslage
Fachinformation
Fachinformationen sind Gebrauchsinformationen über Arzneimittel, die sich an Fachkreise richten. Aufbau und
Inhalt ist im Arzneimittelgesetz (AMG § 11) geregelt. Sie enthalten Angaben zur Zusammensetzung,
Anwendungsgebieten, Dosierungen, Kontraindikationen, Interaktionen, Nebenwirkungen, Notfallmaßnahmen,
pharmakologische Eigenschaften, etc. Der Inhalt wird vom pharmazeutischen Unternehmen erstellt und vom BfArM als
zuständige Behörde für Deutschland freigegeben.
Zugänglich sind sie auf verschiedenen Wegen: fachinfo.de, Seiten der pharmazeutischen Unternehmer, teils als
Service anderer Akteure im Gesundheitswesen.
Bei Markteinführung eines neuen Arzneimittels liegen nur begrenzte klinische Erfahrungen vor. Im günstigsten
Fall/bei größeren
Phase-III-Studien wurden mehrere Tausend Patienten untersucht, so dass – je nach Perspektive „nur“ oder
„immerhin“ - UAW mit einer Häufigkeit bis etwa 0,1 % bei Markteinführung bekannt sein können.
Die genannten UAW selbst sowie deren respektiven Häufigkeiten bilden nicht immer die Realität ab. Auf der einen
Seite ist Prozess der langsam, sodass es sein kann, dass Verdachtsfälle neuer UAW jahrelange nicht aufgeführt
werden (beispielhaft: Restless-Less-Syndrom bei Venlafaxin).[4] Auf der anderen Seite geht es für die
pharmazeutischen Unternehmen auch um rechtliche Absicherung, sodass UAW überbewertet werden können. Das und der
Umstand, dass scheinbar eher die Häufigkeit eines Ereignisses statt einer Auswertung des relativen Risikos
angegeben wird, kann dazu führen, dass Ereignisse als UAW aufgenommen werden, für die wohl eher kein kausaler
Zusammenhang bestehen dürfte (beispielhaft: bei Orlistat (Xenical®) wird „Influenza“ als sehr häufige (betrifft
> 10 % der Patienten) Nebenwirkung angegeben; denn gem. EPAR-Bericht wurde gefunden: Orlistat-Gruppe: 39,7 %;
Placebo-Gruppe: 36,2 %).[1]
Häufigkeitsangaben werden in Kategorien basierend auf einer logarithmischen Skala (> 10 %, 10-1 %, 1-0,1 %,
0,1-0,01 %, < 0,01 %) eingruppiert; hierbei geht Genauigkeit verloren. Ist die Häufigkeit auf Grundlage der
verfügbaren Daten nicht abschätzbar, wird „nicht bekannt“ angegeben. In 909 Fachinformationen oder
vergleichbaren Dokumenten (Prescribing information, EPAR, etc.) werden im Mittel für ca. 22 % (s: 27,5 %)
der UAW die Häufigkeit als „nicht bekannt“ angegeben; beschränkt man sich auf die Fachinformationen mit
mind. 25 UAW (n: 639), ist das Bild vergleichbar (x quer: ca. 19 %; s: 23,8 %).[12] Auf bemerkenswerte Art
schleierhaft können einem dabei manche Fachinformationen sein, bei denen die Häufigkeiten aller UAWs mit
„nicht bekannt“ angegeben wird; z.B. „Aspirin®“, Stand: 09/2019 obwohl „Acetylsalicylsäure vermutlich zu den
am besten erforschten Substanzen überhaupt gehört“.[9]
Pharmakovigilanzdaten
Pharmakovigilanzdaten sind Daten, die aus Spontanmeldungen über unerwünschte Ereignisse generiert werden.
Ein unerwünschtes Ereignis ist noch keine Nebenwirkung, hierzu muss ein Zusammenhang mit der Anwendung
des Mittels bestehen. Spontanmeldungen können sowohl von Ärzten und Apothekern über einschlägige Kanäle
abgegeben werden (respektive AMKen, pharmazeutischer Unternehmer, BfArM), als auch von Patienten.
Spontanmeldungen können ambulante Fälle wie auch Behandlungen im Krankenhaus abdecken. Erfasst wird die
gesamte Patientenpopulation. Die erhobenen Daten eignen sich zur Generierung von Hypothesen über neue
UAW, Indikationen, Eignung bei besonders vulnerablen Gruppen, etc. Sie sind alleine aber noch nicht
beweisend.
Allgemein zugängliche Pharmakovigilanzdaten sind selten noch weiter nach Schwere des gemeldeten
Ereignisses, Dosierung oder Applikationsroute aufgeschlüsselt. Auch können Gründe/Indikation, aus denen
das Mittel angewendet wurde, was Einsicht in weitere Umstände der Therapie geben könnte (z.B. Übelkeit
als AE bei harnstoff-haltiger Pflegecreme; Anwendung im Rahmen eines Zytostatika-induziertem
Hand-Fuß-Syndrom). Auch andere wünschenswerte Informationen zur umfassenderen Beurteilung fehlen oft
(Resultat eines Absetz- oder Reexpositionversuchs, Laborwerte, Komorbiditäten, Latenzzeit bis zum
Auftreten des Ereignisses, etc.).
Die Qualität und Verwendbarkeit steht und fällt damit, wie zuverlässig erkannte unerwünschte Ereignisse
auch tatsächlich gemeldet werden. Bedauerlicherweise ist „under-reporting“ ein großes Problem und
Ereignisse werden aus zahlreichen Gründen nicht gemeldet, u.a. (!): Schuldgefühle gegenüber dem
Patienten, weil er durch die Therapie geschädigt wurde; Zeitmangel zur Meldung; Zaghaftigkeit oder
Angst, sich durch die Meldung als unwissend zu entblößen; fehlende Motivation zur Meldung (UAW bereits
bekannt/durch Meldung keine neue Erkenntnis).[8] Gefunden wurde eine „under-reporting“-Rate von im
Median 94 %.[5]
Damit Häufigkeiten extrapoliert werden könnten, sind in Ermangelung einer Kontrollgruppe neben den
Spontanmeldungen weitere Informationen nötig, wie die Inzidenz des Ereignisses in der nicht-exponierten
Bevölkerung oder wie viele Patienten das Mittel überhaupt anwenden.
Bis 10/2017 wurden offizielle deutsche Daten vom BfArM öffentlich zugänglich gemacht. Ein Teil der Daten
(2005 bis 9/2015) ist weiterhin auf einer Seite von OpenVigil abrufbar.[2, 3] Die Daten sind um
statistische Assoziationen ergänzt, sog. „Disproportionalitäten“; also ein Abweichen der beobachteten
von der erwarteten Häufigkeit. Über die deutschen Daten hinaus sind dort auch die Meldedaten der FDA
zugänglich, bei denen es sich größtenteils um US-amerikanische Daten handelt, zum Teil aber auch
europäische, kanadische usw. Woher die benötigten zusätzlichen Daten kommen, um von Meldungen aus dem
Spontanmeldesystem auf Disproportionalitätsmarker zu extrapolieren, ist für uns unklar, dem Anlagen zum
Projekt nicht zu entnehmen.
Die Daten aus dem europäischen Meldesystem sind einsehbar auf Seiten der EMA/EudraVigilance.[4]
Bereitgestellt werden Rohdaten, einsortiert in vordefinierte Gruppen, z.B. nach Alter, Ereignis, ob die
Meldung von jemanden aus dem Gesundheitswesen abgegeben wurde.
2. Diskussion
Beide Datenpools haben Stärken und Schwächen. Bedauerlicherweise lassen sich die Angaben nicht ineinander
überführen, um die Schwächen eines Systems mit den Stärken des anderen auszugleichen.
Angaben aus Fachinformationen sind Verhältnisse in Form von Häufigkeiten: „Patienten mit Ereignissen
/Patienten insgesamt“. Bei Spontanmeldungen handelt es sich i.d.R. nicht um Verhältnisse, sondern in
Ermanglung einer Placebo-Gruppe um die kontextlose Anzahl der eingegangenen Meldungen: „Patienten mit
Ereignis“ – und diese Anzahl ist wegen „under-reportings“ zu niedrig; wie viel zu niedrig, ist nicht
bekannt und daher nicht direkt kompensierbar, weitere Studien bzw. Analysen wären nötig.
Auf der Internetseite von OpenVigil werden die aktuellen bei der FDA und ein Ausschnitt der beim BfArM
eingegangen Meldungen niedrigschwellig zugänglich gemacht. Hier werden die Meldungsanzahlen ergänzt um
Disproportionalitätsmarker, z.B. die „proportional reporting rate“ (PRR). Die PRR ist das Verhältnis der
Wahrscheinlichkeiten für das Auftreten eines Ereignisses in zwei Gruppen.
Sie ist ein Maß für die Assoziation und soll Aufschluss darüber geben, wie sich exponierte Bevölkerung
zu nicht-exponierter Bevölkerung verhält. Ein Wert von 1 entspricht normalen Hintergrundrauschen, PRR
> 1 spricht für
einen Zusammenhang, PRR < 1 spricht gegen einen Zusammenhang. Ein Anwendungsfall im Alltag kann z.B.
sein, ein Ranking aufzustellen, welcher Arzneistoff für eine beim Patienten bestehende Nebenwirkung
am ehesten ursächlich sein kann.[2]
Mit den drei verschiedenen Messzahlen werden unterschiedliche Fragen beantwortet:
- Fachinformationen
- PRR [OpenVigil]
- Spontanmeldungen
-
"wie viele Personen insgesamt?"
Im Vergleich der Datensätze untereinander lassen sich aber auch leicht Beispiele finden, die dem
pharmazeutisch-medizinischen Sachverstand und der aktuellen Studienlage widersprechen (siehe
Tab. 1).
Tab 1: Gegenüberstellungen von Pharmakovigilanzdaten, Angaben aus Fachinformationen und
Studienlage am Beispiel periphere Ödeme bei Calcium-Antagonisten vom Dihydropyridin-Typ
Gegenüber dem, was als „richtig“ gilt, den Studiendaten der Meta-Analyse, lässt sich weder für
die FDA-Pharmakovigilanzdaten noch für die Angaben aus den Fachinformationen eine höhere
„Genauigkeit“ ableiten.
Die Angaben der EMA mit den reinen Anzahlen der Spontanmeldungen lassen ohne weiteren Kontext
nicht die Beurteilung zu, ob eine Assoziation mit dem Ereignis gegeben ist.
Von den FDA-Daten lässt sich nicht auf die deutsche Bevölkerung schließen.
Verschreibungsverhalten, Bevölkerungsstruktur, Bevölkerungsgesundheit, Pharmakogenetik,
Arzneimittelmarkt, etc. sind zu unterschiedlich.
Orientierend an den Werten der Meta-Analyse ergibt sich folgendes Bild (siehe Tab. 2).
Tab 2: Rating wahrscheinlichster Reihenfolgen für das Auftreten von peripheren Ödem entsprechend
Tab. 1
Gem. FDA-Daten gilt Lercanidipin als wahrscheinlichster Auslöser für periphere Ödeme, gefolgt von
Amlodipin. Das Felodipin- und auch das Nifedipin-CI überschneidet sich mit den anderen
Substanzen. Bei den Fachinformationen ergibt sich ein Bild, das dem Ranking der Meta-Analyse
ähnlicher ist; bei Amlodipin findet sich keine UAW „peripheres Ödem“, verwendet wurde „Ödeme“.
Wegen der Eingliederung in die Häufigkeitskategorien gelten Amlodipin, Nifedipin und Felodipin
(alle „sehr häufig“) als gleich wahrscheinlich, gefolgt von Lercanidipin („häufig“).
Spekulation: Bei Lercanidipin könnte die erhöhte Ödemrate auch mit einer fehlerhaften Einnahme
zusammenhängen. In den Daten wird nicht aufgeschlüsslet, ob die Einnahme nach oder wie es
richtiger wäre, vor der Mahlzeit erfolgt ist.
Das Beispiel ist natürlich ein bisschen Rosinenpickerei und die FDA-Daten von OpenVigil kommen
beim gewählten Beispiel nicht gut weg. OpenVigil-intern gab es für andere Beispiele und
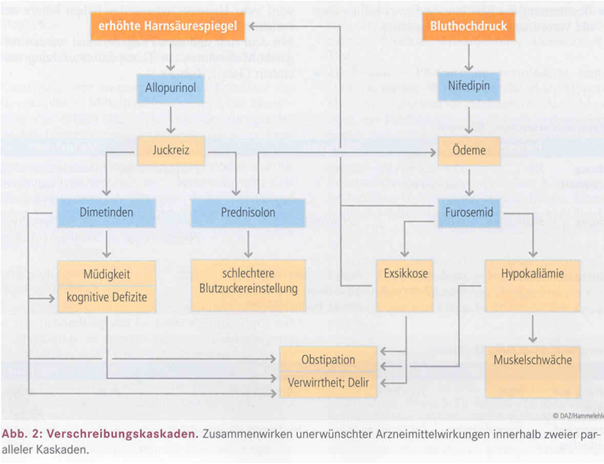
Anwendungsfälle auch positive Tests.[2] Im Kontext von Verschreibungskaskaden haben periphere
Ödeme bei DHP-CCB allerdings eine Art Leuchtturmfunktion und die Zahlen sind wichtig; gefunden
hat man Raten für die Kaskade „auslösendes Mittel: DHP-CCB; folgendes Mittel:
(Schleifen)-Diuretikum“ von 1,4 % (2019) bis 4,6 % (2018).[10, 11]
3. Folgerung
Beide Datenarten, Pharmakovigilanz sowie Fachinformationen, erfordern,
medizinisch-pharmazeutischen Sachverstand walten zu lassen. Auf die Zahlen kann man sich nie
blind verlassen, der Heilberufler muss sie immer auf Plausibilität prüfen.
Die einzigen Pharmakovigilanzdaten mit einer gewissen Aussagekraft sind die um
Disproportionalitätswerte ergänzten der FDA, die sich nur beschränkt auf Deutschland übertragen
lassen. Um die BfArM- oder EMA-Daten verwendbar zu machen, müssten u.a. Inzidenzen zu jedem
Ereignis recherchiert werden, was schlicht unverhältnismäßig ist. Bei den BfArM-Daten stellt
sich zudem die Frage der Aktualität, insgesamt (2005-2015) als auch bezogen auf die damit
einhergehende Wirkstoff-Abdeckung.
Fachinformationen sind objektive Informationsquellen und die angegeben Daten wurden unter
geregelten Bedingungen erhoben. Je nach Produkt ist die Darstellung der Ergebnisse der
klinischen Studien, die bei Zulassung vorgelegt wurden, unterschiedlich nah an den tatsächlich
gefundenen Ergebnissen. Pharmazeutische Unternehmen können dazu neigen, sich einen gewissen
Spielraum einzuräumen, welcher Wert letztlich für die Häufigkeit einer UAW angegeben wird.
Bei Fachinformationen werden die Daten geregelt erhoben und ausgewertet, bei der
Interpretation/Disproportionalitätsanalyse wird sich schließlich Beinfreiheit eingeräumt und
Genauigkeit kann verloren gehen. Bei Pharmakovigilanzdaten wird bei unklarer Ausgangslage
versucht, aus den Daten das meiste herauszuholen.
Fürs Erste wird mit den Daten Fachinformationen gearbeitet und diese mit Angaben aus anderen
belastbaren Quellen, z.B. Studien oder Meta-Analysen, ergänzt, sofern die Daten dort in
Einheiten angegeben werden, die sich übertragen bzw. in den bestehenden Datensatz adäquat
eingliedern lassen.
Perspektivisch kann erwogen werden, die um die PRR ergänzten FDA-Daten parallel anzubieten, wenn
die Herkunft der Daten, die zur Berechnung der PRR ferner nötig sind, geklärt werden kann.
4. Quellen